



服务热线
178 0020 3020
本期文献
1. 肌酸通过激活Smad2/3促进癌症转移【卜鹏程@中国科学院】
2. 羟脂蛋白生物合成增强细胞衰老并检测衰老
3. 骨骼肌中的蛋白酶体应激会产生远程保护性反应,从而延迟视网膜和大脑的衰老
4. 肥胖相关的高瘦素血症改变下丘脑的胶质血管界面以促进高血压
5. 肌动蛋白荧光屏显示,NEMO样激酶是肝脏糖异生的关键抑制因子【李红良@武汉大学】
6. PKM2依赖性肝Th17细胞代谢倾斜调节非酒精性脂肪性肝病的发病机制
7. 代谢灵活性决定了肿瘤微环境中人类NK细胞的功能命运
8. 肿瘤利用FTO介导的糖酵解代谢调节来逃避免疫监视【徐萌@清华大学】
9. 甲硫氨酸-Mettl3-N6-甲基腺苷轴促进多囊肾病

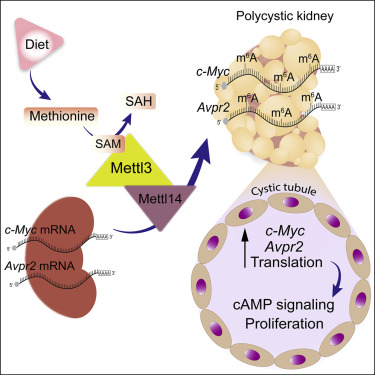
封面:在本期中,Ramalingam等人表明,蛋氨酸补充或RNA甲基转移酶Mettl3的转基因表达可促进肾囊肿的生长,而饮食中蛋氨酸的限制或Mettl3的缺失会减弱这种生长。从机制上讲,他们发现蛋氨酸诱导Mettl3表达,进而通过增强mRNA m6A水平和c-Myc和Avpr2的翻译来促进囊肿生长。该图像是与Dolichos biflorus凝集素(绿色,集合管标记)和磷酸-组蛋白-H3(红色,增殖标记)共染色的小鼠多囊肾的数字增强免疫荧光显微照片。RNA分子交织在收集管和囊肿网络内,表明囊肿加重m6A RNA化学修饰的作用。图片来源:Harini Ramalingam和Vishal Patel。
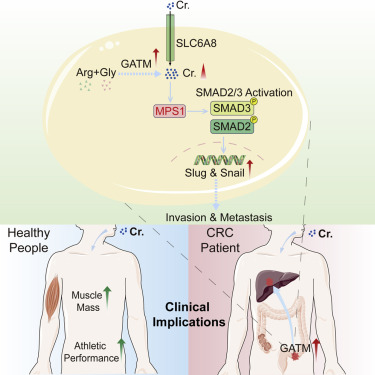
1. 肌酸通过激活Smad2/3促进癌症转移
Creatine promotes cancer metastasis through activation of Smad2/3
作为最受欢迎的营养补充剂之一,肌酸已被高度用于增加肌肉质量和改善运动表现。在这里,我们报告使用原位小鼠模型的肌酸的不利影响,显示肌酸促进结肠直肠癌和乳腺癌转移并缩短小鼠存活。我们显示甘氨酸脒基转移酶(GATM),肌酸合成的限速酶,在肝转移中上调。膳食摄取或GATM介导的肌酸从头合成通过单极纺锤体1(MPS1)激活的Smad2和Smad3磷酸化上调Snail和Slug表达来增强癌症转移并缩短小鼠存活。GATM敲低或MPS1抑制通过下调Snail和Slug来抑制癌症转移并有益于小鼠存活。我们的研究结果要求在考虑膳食肌酸改善肌肉质量或治疗疾病时谨慎使用,并建议靶向GATM或MPS1可预防癌症转移,特别是转化生长因子β受体突变结直肠癌的转移。
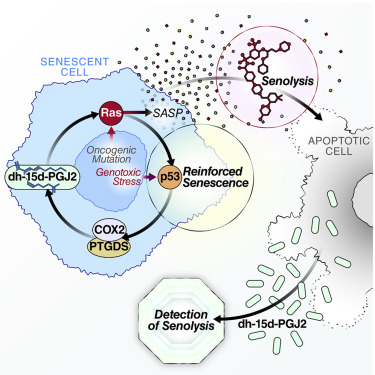
2. 羟脂蛋白生物合成增强细胞衰老并检测衰老
Oxylipin biosynthesis reinforces cellular senescence and allows detection of senolysis
细胞衰老是一种压力或损伤反应,导致永久性增殖性停滞并分泌具有强效生物活性的许多因子。这种衰老相关的分泌表型(SASP)主要用于参与胚胎发生,伤口愈合,炎症和许多与年龄相关的病理学的分泌蛋白。相比之下,SASP的脂质成分未被充分研究。我们显示衰老细胞激活几种oxylipins的生物合成,促进SASP的片段并加强增殖停滞。值得注意的是,衰老细胞合成并积累未研究的细胞内前列腺素1a,1b-二高-15-脱氧-δ-12,14-前列腺素J2。释放的15-脱氧-δ-12,14-前列腺素J2是培养和体内溶解的生物标志物。这种和其他前列腺素D2相关脂质通过激活RAS信号传导促进衰老停滞和SASP。这些数据确定了细胞衰老的一个重要方面和检测衰老的方法。
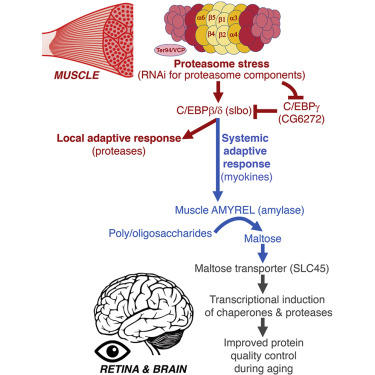
3. 骨骼肌中的蛋白酶体应激会产生远程保护性反应,从而延迟视网膜和大脑的衰老
Proteasome stress in skeletal muscle mounts a long-range protective response that delays retinal and brain aging
中枢神经系统(CNS)中的神经变性具有受外周组织影响的机体衰老的决定性特征。临床观察表明,骨骼肌影响中枢神经系统的衰老,但潜在的肌脑信号仍未得到探索。在果蝇中,我们发现骨骼肌中蛋白酶体的适度扰动会在衰老过程中诱导CNS蛋白稳态的代偿性保存。这种远程应激信号取决于肌肉分泌的淀粉样淀粉酶。模拟肌肉中应激诱导的戊醇上调可通过分子伴侣减少大脑和视网膜中多泛素化蛋白的年龄相关性积累。蛋白抑制的保存源于通过淀粉淀粉酶活性产生的二糖麦芽糖。相应地,SLC45麦芽糖转运蛋白的RNAi降低了戊醇诱导的伴侣蛋白的表达,并在衰老过程中恶化了脑蛋白的稳态。此外,麦芽糖保留了受热应激攻击的人脑类器官中的蛋白质稳态和神经元活性。因此,骨骼肌中的蛋白酶体应激通过淀粉酶/麦芽糖产生适应性反应来阻碍视网膜和大脑衰老。
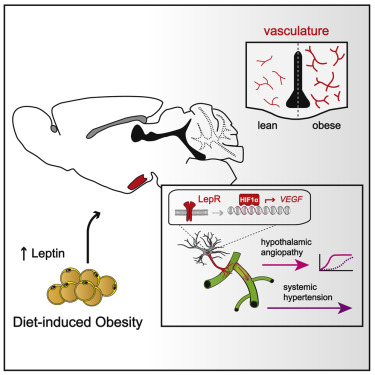
4. 肥胖相关的高瘦素血症改变下丘脑的胶质血管界面以促进高血压
Obesity-associated hyperleptinemia alters the gliovascular interface of the hypothalamus to promote hypertension.
微血管系统和大血管系统的病理学是代谢综合征的标志,其可导致长期升高的血压。然而,所涉及的潜在病理机制仍需要澄清。在这里,我们报告说,肥胖相关的血清瘦素增加触发了调节血液动力学稳态的自主前脑中心微血管构筑的选择性扩展。通过使用一系列细胞和区域特异性损失和功能获得模型,我们表明这种病理生理过程依赖于下丘脑-星形胶质细胞缺氧诱导因子1α-血管内皮生长因子α-瘦素信号传导下游的VEGF)信号传导。重要的是,几种不同的HIF1模型α-星形胶质细胞中的VEGF途径破坏不仅受到肥胖诱导的下丘脑血管病的保护,而且受到交感神经过度活跃或动脉高血压的保护。这些结果表明高瘦素血症通过HIF1促进肥胖诱导的高血压α-VEGF信号级联在下丘脑星形胶质细胞中,同时建立一种新的机制联系,将下丘脑微血管构筑与控制全身血压联系起来。
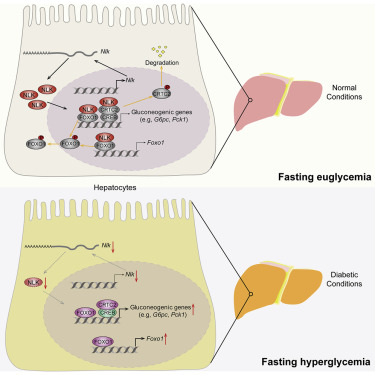
5. 肌动蛋白荧光屏显示,NEMO样激酶是肝脏糖异生的关键抑制因子【李红良@武汉大学】
A kinome screen reveals that Nemo-like kinase is a key suppressor of hepatic gluconeogenesis
抗高血糖治疗是治疗2型糖尿病(T2D)的重要优先事项。过量的肝葡萄糖产生(HGP)是空腹高血糖的主要原因。因此,更好地了解其调节对于开发有效的抗高血糖疗法将是重要的。使用糖异生靶向激酶组筛选方法结合转录组分析,我们发现Nemo样激酶(NLK)作为HGP的有效抑制剂。从机制上讲,NLK磷酸化并促进肝脏糖异生的两个关键调节因子CRTC2和FOXO1的核输出,导致前者的蛋白酶体依赖性降解和抑制自身转录活性和后者的表达。重要的是,NLK的表达在糖尿病个体和糖尿病啮齿动物模型的肝脏中下调,并且在小鼠模型中恢复NLK表达改善了高血糖症。因此,我们的研究结果揭示NLK是糖异生调节网络中的关键参与者,也是T2D的潜在治疗靶点。
6. PKM2依赖性肝Th17细胞代谢倾斜调节非酒精性脂肪性肝病的发病机制
PKM2-dependent metabolic skewing of hepatic Th17 cells regulates pathogenesis of non-alcoholic fatty liver disease
新出现的证据表明Th17细胞对非酒精性脂肪性肝病(NAFLD)发病机制的关键贡献。然而,肝Th17细胞的致病特征和机制仍然未知。在这里,我们揭示和表征足以加剧NAFLD发病机制的炎性肝CXCR3+Th17(ihTh17)细胞的独特群体。肝脏ihTh17细胞应计依赖于肝脏微环境和CXCR3轴激活。机制上,ihTh17细胞的致病潜力与染色质可及性增加,糖酵解输出以及伴随产生的IL-17A,IFNα和IL-17A相关γ, 和肿瘤坏死因子α. 使用2-DG或细胞特异性PKM2缺失调节糖酵解足以逆转以ihTh17为中心的炎症活力和NAFLD严重性。重要的是,ihTh17细胞特征,CXCR3轴激活和糖酵解基因的肝表达在人NAFLD中是保守的。总之,我们的数据显示脂肪肝微环境调节Th17细胞的积累,代谢和对ihTh17命运的能力。这些途径的调节具有开发NAFLD新型治疗策略的潜力。

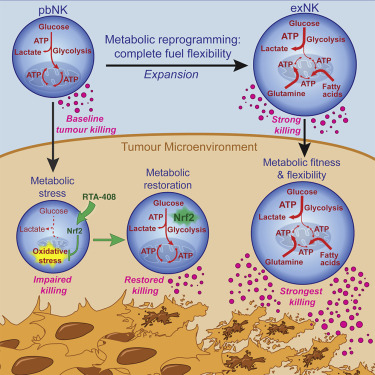
7. 代谢灵活性决定了肿瘤微环境中人类NK细胞的功能命运
Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment
NK细胞是抗肿瘤免疫的核心,并且最近显示出治疗血液恶性肿瘤的功效。然而,它们在恶性肿瘤微环境中的功能障碍仍然是针对实体瘤的癌症免疫疗法的关键屏障。使用癌症患者样本和蛋白质组学,我们发现肿瘤微环境中的人NK细胞功能障碍是由于通过脂质过氧化相关的氧化应激抑制葡萄糖代谢。Nrf2抗氧化途径的激活恢复了NK细胞的代谢和功能,并导致体内更强的抗肿瘤活性。引人注目的是,用完全代谢底物灵活性重新编程的扩增的NK细胞不仅持续代谢适应性,而且矛盾地增加了它们在肿瘤微环境中的肿瘤杀伤和对营养物剥夺的响应。我们的研究结果表明,代谢灵活性使细胞毒性免疫细胞能够利用肿瘤的代谢敌意来获得优势,从而解决癌症免疫治疗的关键障碍。
8.肿瘤利用FTO介导的糖酵解代谢调节来逃避免疫监视【徐萌@清华大学】
Tumors exploit FTO-mediated regulation of glycolytic metabolism to evade immune surveillance
对有助于免疫逃避的因素和调节层的复杂性的不断了解促进了免疫疗法的发展。然而,恶性肿瘤的多样性限制了特定遗传和表观遗传背景下的许多已知机制,表明需要发现一般的驱动基因。在这里,我们已经确定m6A去甲基化酶FTO是肿瘤通过调节糖酵解代谢来逃避免疫监视的必需表位转录组调节剂。我们显示肿瘤细胞中FTO介导的m6A去甲基化提高了转录因子c-Jun,JunB和c/EBPβ, 这允许重新布线糖酵解代谢。Fto敲低损害肿瘤细胞的糖酵解活性,其恢复CD8+T细胞的功能,从而抑制肿瘤生长。此外,我们开发了一种小分子化合物Dac51,它可以抑制FTO的活性,阻断FTO介导的免疫逃避,并与检查点阻断协同作用以更好地控制肿瘤,这表明重新编程RNA表位转录组作为免疫治疗的潜在策略。

9.甲硫氨酸-Mettl3-N6-甲基腺苷轴促进多囊肾病
A methionine-Mettl3-N6-methyladenosine axis promotes polycystic kidney disease
常染色体显性遗传性多囊肾病(ADPKD)是一种常见的单基因疾病,其特征是许多逐渐扩大的肾囊肿。Mettl3是一种催化丰富的N6-甲基腺苷(m6A)RNA修饰的甲基转移酶,与发育有关,但其在大多数疾病中的作用尚不清楚。在这里,我们显示Mettl3和m6A水平在小鼠和人ADPKD样品中增加,并且肾特异性转基因Mettl3表达产生肾小管囊肿。相反,三种直系同源ADPKD小鼠模型中的Mettl3缺失减慢了囊肿的生长。有趣的是,ADPKD模型中蛋氨酸和S-腺苷甲硫氨酸(SAM)水平也升高。此外,蛋氨酸和SAM诱导Mettl3表达并加重离体囊肿生长,而饮食蛋氨酸限制减弱小鼠ADPKD。最后,Mettl3通过增强的c-Myc和Avpr2 mRNA m6A修饰和翻译激活促进囊肿的c-Myc和cAMP途径。因此,Mettl3促进ADPKD并将甲硫氨酸利用与增殖和囊肿生长的表位转录组激活联系起来。

附件