



服务热线
178 0020 3020
本期文章
1. 卵巢癌中细胞成分的单细胞解剖和影响肿瘤免疫表型的相互作用
2. 肿瘤相关巨噬细胞中RNA N6腺苷甲基转移酶Mettl14的缺失促进CD8+T细胞功能障碍和肿瘤生长
3. mRNA上的N6-甲基腺苷促进相分离的核体,其抑制髓样白血病分化
4. 胸腔驻留Tim-4+巨噬细胞损害CD8+T抗肿瘤细胞免疫
5. Durvalumab联合olaparib和紫杉醇治疗高危HER2阴性II/III期乳腺癌:自适应随机I-SPY2试验的结果
6. AML微环境催化逐步演变为吉列替尼耐药

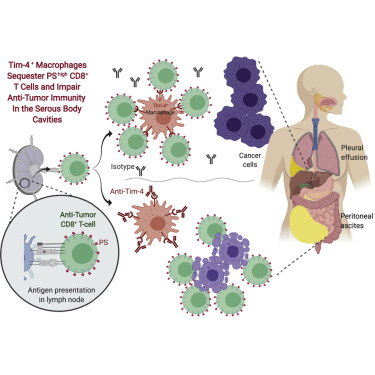
封面:周等人(pp. 973–988) 描述了胸膜腔和腹腔腔中的腔内巨噬细胞,部分通过隔离机制损害抗肿瘤 T 细胞免疫。封面描绘了胸膜腔——肺周围的空间,液体可以在其中积聚。右侧的器官是腹腔内的肝脏。巨噬细胞(蓝色)抑制 T 细胞(绿色)接触肿瘤细胞(洋红色),这可能是由于它们在进化上保守的功能,即限制浆液体腔中的组织损伤。这种限制会损害这些体腔中的抗肿瘤免疫力。
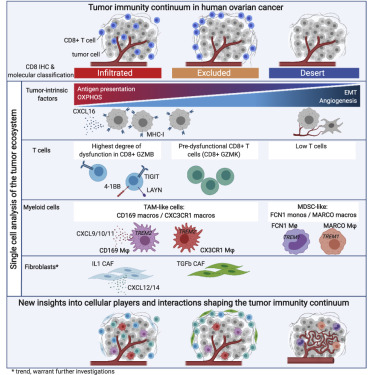
1. 卵巢癌中细胞成分的单细胞解剖和影响肿瘤免疫表型的相互作用
Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer
不同的T细胞浸润模式,即免疫浸润,排除和逃逸,导致对癌症免疫疗法的不同反应。然而,支持这些肿瘤免疫表型的关键决定因素和生物学仍然难以捉摸。在这里,作者通过对15个卵巢肿瘤进行单细胞RNA测序分析,提供了整个肿瘤生态系统的高分辨率解剖。免疫逃逸肿瘤的特征在于独特的肿瘤细胞内在特征,包括代谢途径和低抗原呈递,以及单核细胞和未成熟巨噬细胞的富集。免疫浸润和排除的肿瘤在其T细胞组成和成纤维细胞亚群方面显着不同。此外,作者的研究揭示了隔室内和隔室内趋化因子-受体-配体相互作用作为介导免疫细胞浸润的潜在机制,例如通过CXCL16-CXCR6的肿瘤细胞-T细胞串扰和通过CXCL12/14-CXCR4的基质免疫细胞串扰。作者的数据突出了塑造肿瘤免疫表型的潜在分子机制,并可能为改善癌症免疫疗法的临床益处提供治疗策略。
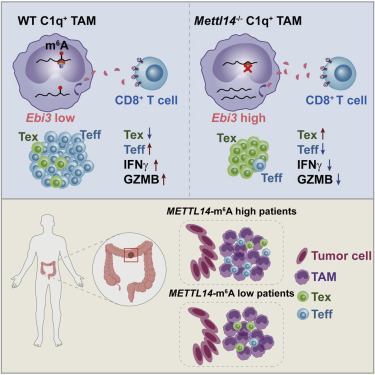
2. 肿瘤相关巨噬细胞中RNA N6腺苷甲基转移酶Mettl14的缺失促进CD8+T细胞功能障碍和肿瘤生长
The loss of RNA N6-adenosine methyltransferase Mettl14 in tumor-associated macrophages promotes CD8+ T cell dysfunction and tumor growth
肿瘤相关巨噬细胞(TAMs)可以抑制T细胞的抗肿瘤活性,但其潜在机制仍未完全了解。在这里,作者显示C1q+TAMs受RNA N6-甲基腺苷(m6A)程序调节,并通过表达多种免疫调节配体调节肿瘤浸润性CD8+T细胞。巨噬细胞特异性敲除m6A甲基转移酶Mettl14沿着功能失调的轨迹驱动CD8+T细胞分化,损害CD8+T细胞以消除肿瘤。Mettl14缺陷的C1q+TAM显示细胞因子亚基Ebi3的m6A丰度降低和转录物水平升高。此外,EBI3的中和导致功能失调的CD8+T细胞的再活化并克服小鼠中的免疫抑制作用。作者显示METTL14-m6A水平与结直肠癌患者中功能失调的T细胞水平呈负相关,支持该调节途径的临床相关性。因此,作者的研究证明了TAM中的m6A甲基转移酶如何促进CD8+T细胞功能障碍和肿瘤进展。
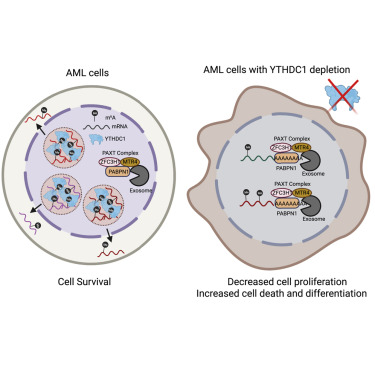
3. mRNA上的N6-甲基腺苷促进相分离的核体,其抑制髓样白血病分化
N6-Methyladenosine on mRNA facilitates a phase-separated nuclear body that suppresses myeloid leukemic differentiation
mRNA上的N6-甲基腺苷(m6A)介导不同的生物过程,其失调导致肿瘤发生。m6A如何决定其在白血病中的多种分子和细胞效应仍然未知。作者发现YTHDC1是来自全基因组CRISPR筛选的髓样白血病中必需的m6A阅读器,并且m6A是YTHDC1进行液-液相分离并形成核YTHDC1-m6A缩合物(NYAC)所必需的。与正常造血干细胞和祖细胞相比,急性髓性白血病(AML)细胞中NYAC的数量增加。AML细胞需要NYAC维持细胞存活和对白血病维持至关重要的未分化状态。此外,NYAC使YTHDC1能够保护m6A mRNA免受PAXT复合物和外来体相关RNA降解的影响。总的来说,m6A是形成由相分离介导的核体所必需的,其维持mRNA稳定性并控制癌细胞存活和分化。
4. Tim-4+腔驻留巨噬细胞损害抗肿瘤CD8+T细胞免疫
Tim-4+ cavity-resident macrophages impair anti-tumor CD8+ T cell immunity
免疫检查点封锁(ICB)一直是癌症的显着临床进展;然而,大多数患者对ICB治疗没有反应。作者显示胸膜腔和腹膜腔中的转移性疾病与ICB治疗后的不良临床结果相关。腔内巨噬细胞表达高水平的磷脂酰丝氨酸(PS)受体Tim-4,这与癌症患者胸腔积液和腹膜腹水中具有肿瘤反应特征的CD8+T细胞数量减少有关。作者证明存活的和细胞毒性的抗肿瘤CD8+T细胞上调PS,这使得它们易于隔离远离肿瘤靶标和Tim-4+巨噬细胞的增殖抑制。Tim-4阻断消除了这种隔离和增殖抑制,并增强了小鼠抗PD-1治疗和过继性T细胞治疗模型中的抗肿瘤功效。因此,Tim-4+腔驻留巨噬细胞限制了免疫疗法在这些微环境中的功效。
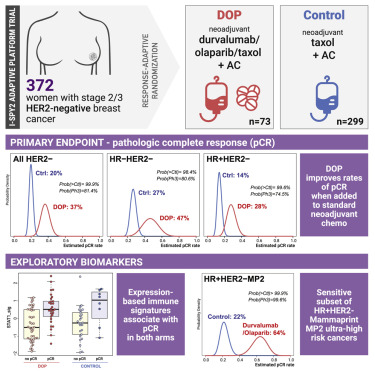
5. Durvalumab联合olaparib和紫杉醇治疗高危HER2阴性II/III期乳腺癌:自适应随机I-SPY2试验的结果
Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: Results from the adaptively randomized I-SPY2 trial
在II/III期HER2阴性乳腺癌的II期I-SPY2试验中研究了加入标准紫杉醇新辅助化疗(durvalumab/olaparib/紫杉醇[DOP])的PD-L1抑制剂durvalumab和PARP抑制剂olaparib的组合。73名参与者被随机分配到DOP和299名标准护理(紫杉醇)对照。DOP增加了所有HER2阴性(20%-37%),激素受体(HR)阳性/HER2阴性(14%-28%)和三阴性乳腺癌(TNBC)的病理完全缓解率(pCR)(27%-47%)。在HR阳性/HER2阴性癌症中,MammaPrint超高(MP2)病例从DOP中选择性受益(pCR 64%对22%),MP1癌症没有获益(pCR 9%对10%)。总体而言,DOP组中12.3%的患者出现免疫相关的3级不良事件,而对照组为1.3%。与免疫应答相关的基因表达特征与两组中的pCR正相关,而肥大细胞特征与非pCR相关。DOP在HER2阴性乳腺癌中具有优于标准新辅助化疗的功效,特别是在高风险HR阳性/HER2阴性患者的高度敏感亚组中。
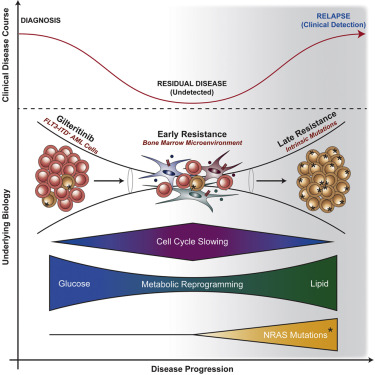
6. AML微环境催化逐步演变为吉列替尼耐药
The AML microenvironment catalyzes a stepwise evolution to gilteritinib resistance
作者的研究详细描述了FLT3突变的急性髓性白血病(AML)中吉列替尼耐药的逐步演变。早期抗性由骨髓微环境介导,其保护残留的白血病细胞。随着时间的推移,白血病细胞进化出内在的抗性机制或晚期抗性。作者通过整合全外显子组测序,CRISPR-Cas9,代谢组学,蛋白质组学和药理学方法来机械地定义早期和晚期抗性。早期耐药细胞经历代谢重编程,生长更慢,并依赖于极光激酶B(AURKB)。晚期耐药细胞的特征在于预先存在的NRAS突变亚克隆的扩增和持续的代谢重编程。作者的模型密切反映了用吉列替尼治疗的AML患者的时间和突变。AURKB的药理学抑制使来自吉列替尼治疗的AML患者的早期抗性细胞培养物和原代白血病细胞重新敏感。这些发现支持在预先存在的抗性突变发生之前用AURKB抑制剂和吉列替尼靶向早期抗性AML细胞的组合策略。
本期链接:https://www.cell.com/cancer-cell/issue?pii=S1535-6108(20)X0008-0#

附件