



服务热线
178 0020 3020
本期文献
1. CD155/TIGIT 轴促进和维持表达新抗原的胰腺癌的免疫逃避
2. 激活协同先天适应性免疫反应以控制转移
3. MAPK抑制剂组合前的抗PD-1/L1导入可最大限度地提高抗肿瘤免疫力和疗效
4. 同时靶向 TGF-β/PD-L1 通过重编程肿瘤微环境克服免疫逃避与放疗协同作用
5. 使用 CDK2/4/6 抑制剂扩大对肿瘤细胞周期的控制
6. 弥漫性大 B 细胞淋巴瘤中肿瘤细胞状态和生态系统的景观

封面:Sun 等人。(pp. 1361–1374) 描述了一种通过协作的先天适应性免疫反应来控制转移的方法。封面描绘了由单磷酰脂质 A (MPLA) 和干扰素 γ (IFNγ) 治疗诱导的从促进肿瘤(左)到抑制肿瘤(右)的微环境变化。MPLA+IFNγ 将肿瘤相关巨噬细胞(皱褶/不规则形状)重新编程为杀伤肿瘤的巨噬细胞,并激活细胞毒性 T 细胞(圆形)以杀死癌细胞(椭圆形)。结果是局部肿瘤控制和抑制转移的全身免疫反应。胡安·曼努埃尔·加西亚·卡塞雷斯的封面艺术作品。
1. CD155/TIGIT 轴促进和维持表达新抗原的胰腺癌的免疫逃避
The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer
CD155/TIGIT 轴可以在慢性病毒感染和癌症的免疫逃避过程中被选择。胰腺癌 (PDAC) 是一种高度致命的恶性肿瘤,迄今为止,以免疫为基础的抗击这种疾病的策略基本上没有成功。我们证实了先前的报告,即大部分 PDAC 具有预测高亲和力 MHC I 类限制性新表位,并将这些发现扩展到晚期/转移性疾病。使用表达新抗原的 PDAC 的多个临床前模型,我们证明了肿瘤内新抗原特异性 CD8 +T 细胞采用多种功能障碍状态,类似于 PDAC 患者的肿瘤浸润淋巴细胞。从机制上讲,CD155/TIGIT 轴的遗传和/或药理学调节足以促进表达本土新抗原的 PDAC 的免疫逃避。最后,我们证明了 CD155/TIGIT 轴对于维持 PDAC 中的免疫逃避至关重要,并揭示了一种联合免疫疗法(TIGIT/PD-1 联合阻断加 CD40 激动)在临床前模型中引发了深刻的抗肿瘤反应,现在准备好临床评价。

2. 激活协同先天适应性免疫反应以控制转移
Activating a collaborative innate-adaptive immune response to control metastasis
肿瘤相关巨噬细胞 (TAM) 促进转移并抑制 T 细胞,但巨噬细胞可以极化以杀死癌细胞。因此,巨噬细胞极化可能是控制癌症的一种策略。我们表明来自乳腺癌患者转移性胸腔积液的巨噬细胞可以被极化以用单磷酰脂质 A (MPLA) 和干扰素 (IFN) γ 杀死癌细胞。MPLA + IFNγ 瘤内或腹膜内注射可减少乳腺癌小鼠模型中原发肿瘤的生长和转移,抑制转移,并增强卵巢癌模型中的化疗反应。巨噬细胞和 T 细胞对于治疗的抗转移作用至关重要。MPLA + IFNγ 刺激 I 型 IFN 信号传导,重编程 CD206 +TAM 诱导 NO 合酶 (iNOS) +巨噬细胞,并通过巨噬细胞分泌的白细胞介素 12 (IL-12) 和肿瘤坏死因子 α (TNFα) 激活细胞毒性 T 细胞。MPLA 和 IFNγ 在临床实践中单独使用,共同代表了一种以前未探索过的参与全身性抗肿瘤免疫反应的方法。
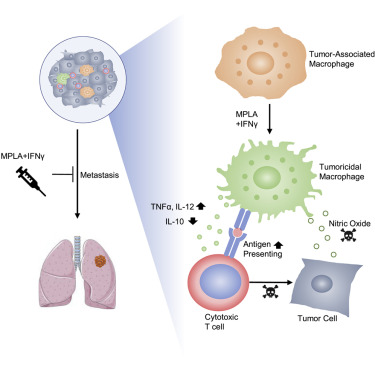
3. MAPK抑制剂组合前的抗PD-1/L1导入可最大限度地提高抗肿瘤免疫力和疗效
Anti-PD-1/L1 lead-in before MAPK inhibitor combination maximizes antitumor immunity and efficacy
合理测序并结合 PD-1/L1 和 MAPK 靶向治疗可能会克服先天性和获得性耐药性。由于 MAPK 抑制剂 (MAPKi) 的临床获益增加与之前的免疫检查点治疗相关,我们比较了连续和/或组合方案在由Braf V600、Nras或Nf1突变以及结直肠和由Kras G12C驱动的胰腺癌。MAPKi 组合之前的抗 PD-1/L1 导入通过促进巨噬细胞的促炎极化和干扰素-γ hi和 CD8 + 的克隆扩增来优化反应持久性高表达激活基因的细胞毒性和增殖性(相对于 CD4 +调节性)T 细胞。由于黑色素瘤脑转移 (MBM) 的治疗耐药性限制了患者的存活率,我们证明在 MAPKi 组合之前测序抗 PD-1/L1 治疗可抑制 MBM 并通过颅内和颅外转移部位的强大 T 细胞克隆扩增提高小鼠存活率。我们建议在 MAPKi 联合治疗前临床测试短暂的抗 PD-1/L1(± 抗 CTLA-4)剂量以抑制治疗耐药性。
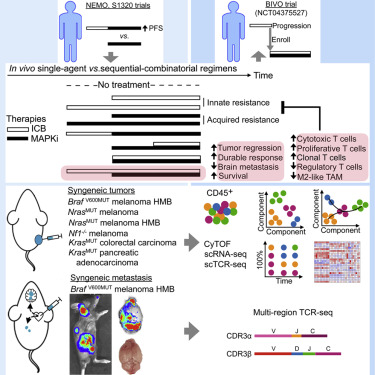
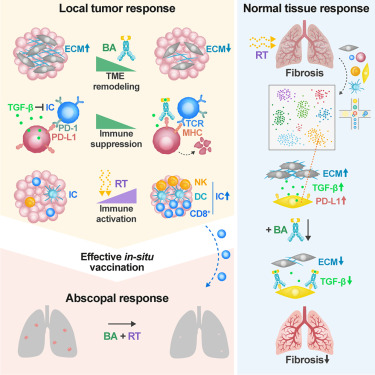
4. 同时靶向 TGF-β/PD-L1 通过重编程肿瘤微环境克服免疫逃避与放疗协同作用
Simultaneous targeting of TGF-β/PD-L1 synergizes with radiotherapy by reprogramming the tumor microenvironment to overcome immune evasion
局部放疗 (RT) 诱导免疫原性抗肿瘤反应,部分被免疫逃避和组织重塑过程的激活所抵消,例如,通过程序性细胞死亡配体 1 (PD-L1) 和转化生长因子 β (TGF-β) 的上调)。我们报告了一种同时抑制 TGF-β 和 PD-L1 的双功能融合蛋白 bintrafusp alfa (BA),可有效地与放疗协同作用,从而在免疫浸润较差的多种抗药性小鼠肿瘤模型中获得更高的存活率。BA + RT (BART) 组合增加肿瘤浸润的白细胞,重新编程肿瘤微环境,并减弱 RT 诱导的纤维化,导致肿瘤免疫重建和自发性肺转移的消退。一贯地,+ T 细胞。有趣的是,BA将 TGF-β 陷阱靶向 PD-L1 +内皮和 M2/脂肪成纤维细胞样细胞区室,减弱了晚期 RT 诱导的肺纤维化。总之,结果表明 BART 组合有可能根除耐药肿瘤,同时保留正常组织,进一步支持其临床转化。
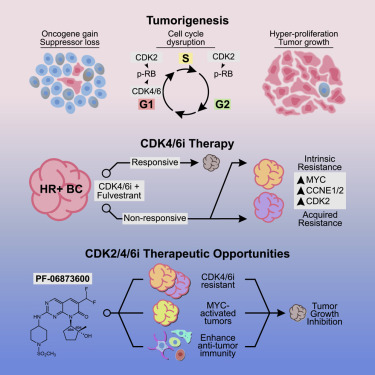
5. 使用 CDK2/4/6 抑制剂扩大对肿瘤细胞周期的控制
Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor
CDK4/6 抑制剂 palbociclib (PAL)与抗激素药物联合使用时,可显着提高 HR + /HER2 -乳腺癌的无进展生存期。我们试图在临床前模型中并通过对临床转录组样本的分析来发现 PAL 耐药机制,这些机制在MYC癌基因和细胞周期蛋白 E/CDK2 活性的诱导上结合。我们建议用小分子靶向 G 1激酶 CDK2、CDK4 和 CDK6 克服对 CDK4/6 抑制的抵抗。我们描述了 PF-06873600 (PF3600) 的药效学和功效,PF-06873600 是一种吡啶并嘧啶,在多个体内有效抑制 CDK2/4/6 活性和功效。 肿瘤模型。连同临床分析,MYC活性预测 (PF3600) 跨多个细胞谱系的功效。最后,我们发现 CDK2/4/6 抑制不会损害同基因模型中的肿瘤特异性免疫检查点阻断反应。我们预计 (PF3600) 目前处于 1 期临床试验阶段,可为 CDK4/6 抑制不足以改变疾病进展的癌症患者提供治疗选择。
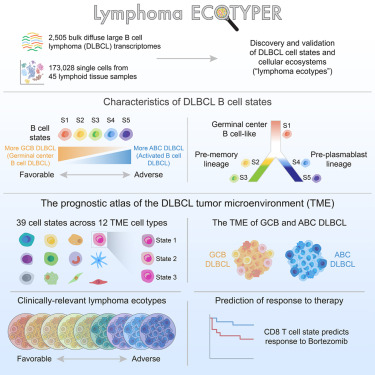
6. 弥漫性大 B 细胞淋巴瘤中肿瘤细胞状态和生态系统的景观
The landscape of tumor cell states and ecosystems in diffuse large B cell lymphoma
弥漫性大 B 细胞淋巴瘤 (DLBCL) 的生物学异质性部分由细胞源亚型和相关基因组病变驱动,但也由肿瘤微环境 (TME) 中的不同细胞类型和细胞状态驱动。然而,大规模剖析这些细胞状态及其临床相关性仍然具有挑战性。在这里,我们实施了 EcoTyper,这是一种集成转录组解卷积和单细胞 RNA 测序的机器学习框架,以表征临床相关的 DLBCL 细胞状态和生态系统。使用这种方法,我们确定了恶性 B 细胞的五种细胞状态,它们的预后关联和分化状态各不相同。我们还确定了其他 12 个谱系的细胞状态的显着变化,这些谱系包括 TME,并在刻板的生态系统中形成细胞状态相互作用。虽然起源细胞亚型具有不同的 TME 组成,但 DLBCL 生态系统捕获现有亚型内的临床异质性,并扩展到起源细胞和基因型类别之外。这些结果以系统级分辨率解决了 DLBCL 微环境,并确定了治疗靶向的机会。
https://www.cell.com/cancer-cell/issue?pii=S1535-6108(20)X0011-0

附件